
第66期
出刊日:2023-10-13
返回上一層基因治療時代來臨! 健保給付天價藥 改變脊髓肌肉萎縮病兒命運
罹患脊髓肌肉萎縮症的孩子,有些可能活不過半年,有些活下來了卻無法坐、立或站起,注定一輩子坐輪椅;但因為基因治療藥物問世,且健保近期納入給付,病人的命運獲得翻轉!
諮詢╱簡穎秀(臺大醫學院臨床教授、臺大醫院基因醫學部主任)
撰稿╱黃筱珮
健保署今(2023)年8月納入給付脊髓性肌肉萎縮症(spinal muscular atrophy, SMA)的基因治療藥物「諾健生」(Zolgensma),經議價後一劑仍然高達4900萬元,成為台灣健保史上最貴的給付藥物,然而這款藥物施打後終生有效,可一舉扭轉患者命運。健保採分期付款、並設有「出生6個月內發病且帶有基因突變」的給付門檻,預估一年有8位病童受惠。
諾健生問市後,在許多國家都有成功治療SMA的案例,加拿大亞伯達省政府曾於2020年資助罹患第二型SMA的2歲男童Mighty Max接受治療,Mighty Max的母親向媒體表示,若非接受治療,Mighty Max可能永遠只坐在輪椅上,不可能獨自行走或站立,治療後肌力明顯增強,能夠獨立完成很多事,看到這些改變,內心充滿感謝。
SMA分四型 第一型最嚴重
脊髓性肌肉萎縮症是一種體隱性遺傳罕見疾病,因為運動神經元存活基因(SMN1)缺損,無法正常製造維持運動神經元健康的SMN蛋白,影響運動神經元的表現,嚴重者可能導致脊髓及腦幹運動神經元壞死,引起持續性的肌肉萎縮、無力,最終發生行走或進食困難,甚至無法呼吸而危及生命。
SMA依發病時間及疾病嚴重程度分為四型,第一型最嚴重,在出生半年內就會發病,無法坐立,爬和站更是奢求,呼吸與吞嚥功能也明顯受損,在未有藥物可治療的年代,通常兩歲前就會死亡。這也是此次健保給付基因治療藥物的對象。
第二型SMA通常在出生後7~18個月發病,病童可坐但無法站立,通常需要一直坐輪椅,會產生很嚴重的脊椎側彎,呼吸功能受到影響,可能常因肺炎住院,若是沒有很好的照顧、營養支持與復健,也容易有生命危險。
第三型則在兒童時期會行走後才發病,患者因運動神經元逐漸退化而使四肢運動功能受損,無法跑步、跳躍。
第四型則在18歲成年以後才發病,症狀較輕,但發病後也可能影響行動能力。
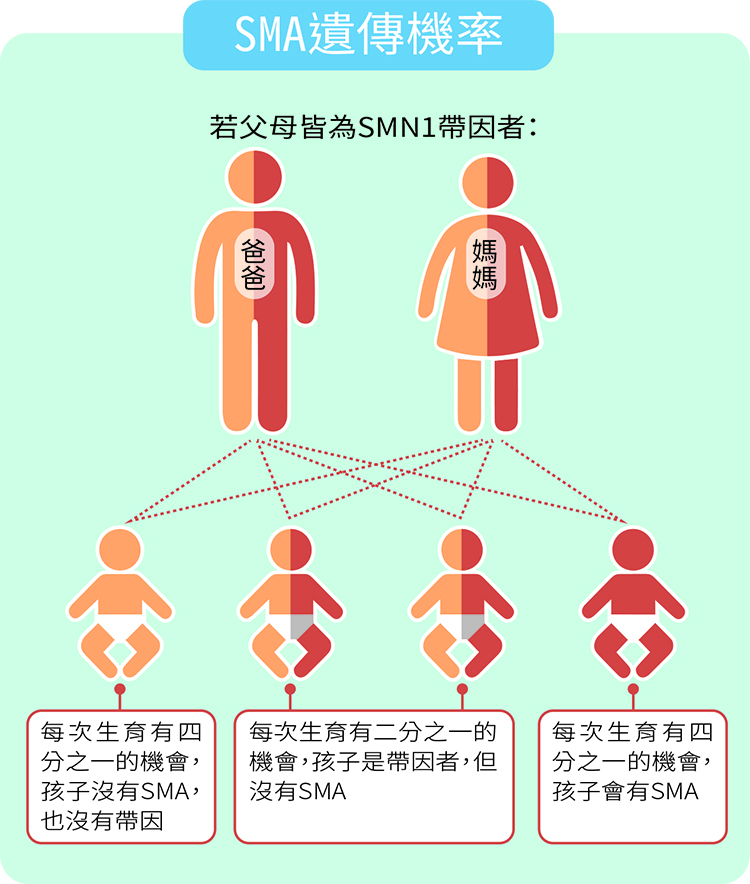
統計顯示國人SMA的帶因機率相當高,約為五十分之一,若不巧父母皆為帶因者,則每一胎不管男孩或女孩,皆會有四分之一的機會成為患者,整體發生率約為萬分之一。
若依目前國內每年出生的新生兒數13萬人口計算,每年約會新增13名SMA患者;不過因為很多新婚夫妻或孕產婦會在孕前或婚前接受SMA基因篩檢,整體發生率已降至三萬分之一。而國內目前已知有就診紀錄的病友約400人。
決定病情輕重關鍵在SMN2基因
同樣是SMN1基因缺損致病,卻有一至四型的差別,為何如此?主要的決定關鍵在於SMN1的「備胎基因」,稱為SMN2。
人體非常奧妙,一般人的SMN1基因數量從一套到三套均有,即便只有一套SMN1基因(即帶因者),也足以支持運動神經元存活,但SMA病人的SMN1基因數量是「0」,所以必須相對依賴SMN2這個備胎基因。
不過SMN2僅有SMN1約十分之一的功能,而SMN2的數量從0到10套都有,所以對體內缺乏SMN1基因的病人來說,SMN2的基因套數愈多,症狀就愈輕微,反之症狀就愈嚴重。
2016年首支SMA藥物問世
突破無藥可治困境
過去針對SMA病人僅能給予支持性療法,提供復健、營養與呼吸照護等支持,這種無藥可治的情況直到2016年第一個SMA治療藥物脊瑞拉注射液(Spinraza)問世,才有了改變。
Spinraza上市時被稱為「奇蹟之藥」,每4個月施打一劑,經由病人背部打入脊髓液裡面,能減少運動神經元壞死。治療機轉是結合SMN2的基因剪接位點,使SMN2也可產出更多正常長度且具有功能的SMN蛋白,大幅提高患者存活率,對病人的肌肉萎縮症狀以及罹病嬰兒的動作發展都有顯著進步。
2014年臺大醫院參與的一項全球性臨床試驗,25位在症狀開始前就接受Spinraza治療的SMA嬰兒至今都存活,狀況十分良好,只有一位孩子需要使用夜用型呼吸器。該項追蹤研究也證實,只要在運動神經元未退化、肌肉關節未攣縮前啟動治療,效果就會非常顯著。
緊接著問世的第二種SMA藥物—服脊立口服溶液用粉劑(Evrysdi),也是修飾SMN2基因剪接(splicing modifier)的藥物,每天服用能提升或維持SMN蛋白濃度,達到治療效果。
上述藥物均有健保給付,健保並於2023年4月起放寬給付範圍,符合「3歲內發病確診:起始治療年齡未滿7歲,或起始治療年齡滿7歲或以上,且上肢運動評估RULM≧15」條件的病友,可由健保給付治療藥物。
圖說:諾健生(Zolgensma)為SMA的一次性基因治療藥物。
一次性基因療法帶來根治希望
台灣有8~10位SMA病童治療成功
在上述兩種藥物之後,一次性的基因治療藥物「諾健生」問世,更為患者帶來無限希望。不同於上述兩種藥物的治療機轉,它是利用容易到達運動神經元的「腺相關病毒載體」攜帶正常的SMN1基因,注射至患者體內,直接填補患者缺乏的SMN1基因,達到「治本」的效果。
目前健保通過的給付條件為「出生6個月內發病且帶有SMN1基因變異」的SMA患者,也就是比較嚴重的第一型,給付價格為一劑4900萬,預計每年約8名病童受惠。
臺大醫院曾在2017年參與諾健生的臨床試驗及恩慈療法,優先用於治療嚴重的第一型嬰兒,打破了過去沒有藥物之前多半活不過兩歲的殘酷現實。同樣的,諾健生的一個全球性臨床試驗,針對發病前的孩子給予治療,這些孩子目前狀況良好,都可以自主呼吸,運動功能也與一般孩子差不多,特別是有3套SMN2基因的孩子反應更佳,證實了藥物有效,只要及早治療,效果顯著。
新生兒篩檢 有助及早發現SMA
如何知道新生兒是否有SMA等疾病?國民健康署自108年擴增補助先天性甲狀腺低能症、苯酮尿症等21項新生兒篩檢項目,新生兒篩檢中心另提供有12項包括SMA在內的「自費」篩檢項目,只要新生兒出生48小時內採取微量腳跟血,就可以完成篩檢。
新生兒篩檢SMA的方式是檢測SMN1基因數目,不過檢驗的準確率非百分百,約2%至5%的患者SMN1基因數目正常但「功能」不正常,無法篩檢出來,另外也可能因為有4套或更多的SMN2基因,所以病情較輕微,需要長期追蹤才能及早偵測發病時機。
產前檢查可自費驗SMA基因
唯檢查前應做好心理準備
至於產前檢查,SMA基因篩檢也是自費項目,有些女性是在懷孕前就自費接受SMA基因檢查,或與另一半攜手接受婚前健檢,及早了解自己和另一半是否為SMA的帶因者,做好準備。
有些婦女則於懷孕初期選擇做檢查,然而,檢查之前建議尋求專業遺傳諮詢師的協助,預先思考檢測結果可能帶來的衝擊,才不會措手不及。因為若檢查出自己為SMA帶因,且先生也帶因,那就有四分之一的機會生下SMA寶寶,甚至也曾有個案因此檢查出自己沒有SMN1基因但未發病,未來可能成為發病而行動能力逐漸退化的SMA病人。有的受檢者得知結果後晴天霹靂,難以接受;還有孕婦發現自己和先生都是SMA帶因者,躊躇著該不該生下孩子,內心也非常折磨。即使做好準備進行了帶因檢查,檢驗的侷限性例如可能有偽陰性或是寶寶自己本身發生基因變異,也需要事先提醒,方能及時於新生兒時期加做篩檢或是於寶寶出現異樣時,儘速再進行相關診斷檢查。
不過,不論檢查結果如何,SMA現在已非讓人束手無策的無解罕病,建議病友懷抱信心,保持樂觀心態,相信隨著基因治療藥物研究如火如荼發展,明天一定會比今天更好。
BOX新生兒篩檢公費項目盼盡快更新
少子化時代,每個小孩都是寶,不過攸關是否能及早發現異常及早治療的新生兒篩檢卻有一些隱憂。衛福部國民健康署目前補助的新生兒篩檢項目共21項(政府補助200元,還要自費約500~1200元),是早在2002年就開始以自費篩檢方式評估篩檢效益的項目,後來又有一些新的疾病篩檢項目如脊髓肌肉萎縮症、龐貝氏症、黏多醣症、嚴重複合型免疫缺乏症等開發出來,為保障寶寶及早獲得診斷與治療的機會,只能先以自費方式提供家長選做,這部分可能因為檢驗費用較高或是因為非政府補助項目公信力不足,或是因為會影響寶寶的商業保險投保資格,不是每個家庭都有意願進行篩檢,形成疾病防治的漏洞,有必要盡快納入公費項目,強化篩檢面向。
請別錯過本期其他精彩內容…
- 編輯筆記:醫學尖端療法 盼扭轉癌症與重大疾病
- 張學友演唱會跌坐舞台 梅尼爾氏症不只會眩暈 也可能喪失聽力
- 藝人自曝4年長12顆息肉! 定期糞便潛血與大腸鏡檢查 遠離大腸癌
- CAR-T殲滅癌細胞奏效 其他細胞治療也是治癌的新希望?
- 細胞療法 可治癒第1型糖尿病?!
- 人工智慧結合超音波 15分鐘「快篩」 揪出阻塞性睡眠呼吸中止症
- 精準投彈! ADC抗體藥物複合體 抗癌精準利器
- 基因治療時代來臨! 血友病可望打一針治癒
- 減重方式愈來愈多 免開腹也能縮胃 「瘦瘦針」新藥輪番上市
- 不只小孩要打疫苗 成人疫苗你接種了嗎?
- 毒品可用來治療憂鬱症?! 擺脫憂鬱還可以怎麼做?
- 扁桃腺發炎 嚴重可能需要插管、住加護病房?!
- 愛挖鼻孔, 小心鼻前庭炎,甚至影響腦部!
- 醫界翻修冠心病指引 不再稱「穩定性」冠狀動脈疾病 強調預防更重於治療
- 阿斯巴甜列可能致癌物, 可以安心吃嗎?
- 楊智超醫師專欄:跑步時,怎麼手腳麻麻的? 運動及健身所引起的周邊神經病變
- 消脂保肝專欄:脂肪也會累積在胰臟! 與肥胖、代謝症候群、糖尿病前期密切相關
- 王清泓醫師專欄:如何陪伴患有青光眼的家人?