
第67期
出刊日:2024-01-15
返回上一層基因性視網膜退化有失明危機 基因治療重見光明!
有一些人因遺傳或基因突變,在青壯年時期甚至幼兒時期就開始視力不良,出現雙眼對稱性的視力與視野喪失,所幸,眼科基因治療已有成功案例,為這群原本注定失明的病人打開新視野,邁向光明人生!
諮詢╱陳達慶(臺大醫學院眼科臨床助理教授、臺大醫院眼科部主治醫師 )
撰稿╱黃筱珮
國內有上萬名「基因性視網膜退化疾病」患者,也稱為「遺傳性視網膜失養症」(inherited retinal degeneration, IRD),這類基因缺陷造成的先天性視力疾病過去被認為無法治療,連病理機轉都不易釐清,醫病雙方面對此類疾病都束手無策。
近年來基因醫學進步,為IRD疾病的診斷與治療帶來巨大改變,國內已有IRD患者經由基因治療明顯好轉,展開全然不同的人生!此療法目前正在申請健保給付中,期待更多患者能夠受惠,順利揮別黑暗,迎向光明。
至少與270個基因缺陷有關
IRD是一群疾病的總稱,大多屬於單基因疾病,可以是顯性、隱性或性聯遺傳。根據美國視網膜資訊網絡(Retinal Information Network, retNet)資料庫,目前已知有超過270個基因缺陷可造成此類疾病,不同基因缺陷引起的狀況也不太一樣,可造成視網膜感光細胞(photoreceptors)與視網膜色素上皮細胞(retinal pigment epithelium, RPE)的原發或次級退化,使視力受損甚至失明,估計台灣有12,000至15,000名患者,並不如想像中少見。例如較為人熟知的「夜盲症」,以及部分已有基因治療的萊伯氏先天性黑矇症(Leber congenital amaurosis, LCA)。
由於IRD疾病牽涉的基因種類廣泛加上表現型分歧,例如同樣是夜盲症,有的早早發病、有的很晚才發病,症狀更是輕重有別,在過去基因醫學尚不發達的年代,臨床醫師要做病程預測、遺傳諮詢、甚或擬定治療方針都有難度,面對病人詢問:「會不會遺傳?能不能生小孩?」「我什麼時候會失明?」醫師也無法找到答案。
幸而近年來次世代基因定序(NGS)技術的突破,讓基因診斷便利性大增,逐一揭開IRD疾病的密碼,加上非侵入性眼底影像檢查的進步與AI智慧運算病程分析,以及以腺相關病毒(AAV)為平台的基因治療藥物開發,連帶使IRD診斷與治療都有大幅進展。
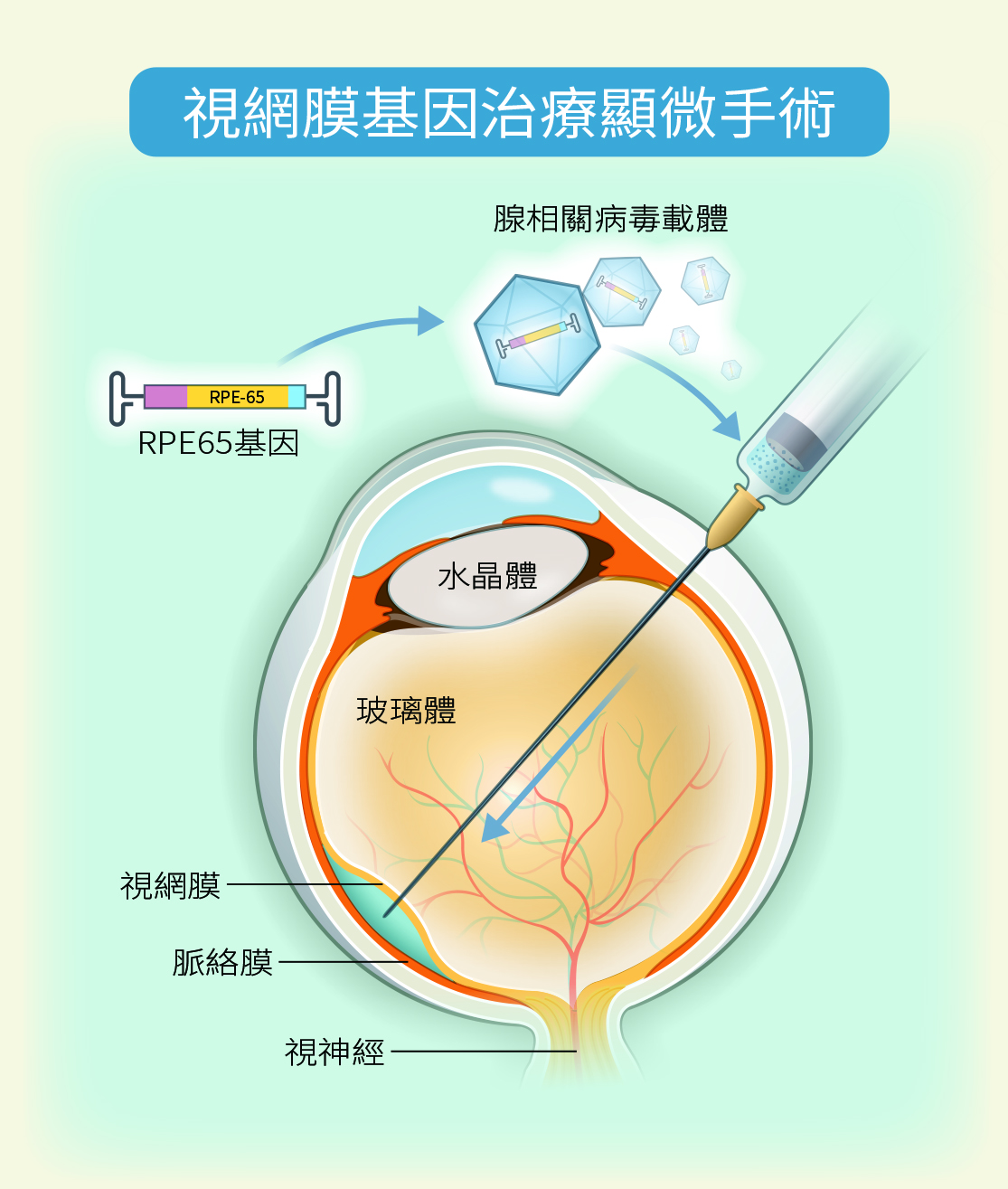
無解眼疾迎來基因治療時代 男學生晚上終於敢出門了
2021年5月,臺大醫院完成東亞首例視網膜基因治療顯微手術,受術患者是一名17歲的高中男學生,他3歲即被診斷出罹患罕見的萊伯氏先天性黑矇症(LCA),平時白天視力還好,所以可以正常就學,但是到了晚上視力就變得很差,完全不能出門,還有視野縮小的症狀,但一直都無法治療。NGS基因定序問世後,確診他是RPE65基因缺陷之LCA,隨著時間視力會日趨惡化。
當時國外已有LCA病人基因治療的成功案例,但亞洲尚無類似的案例,加上新興治療的費用昂貴,需要有實例證明療效,後續較有可能促成健保給付,醫療團隊評估男學生中心視力僅剩小範圍,再拖延恐怕病況惡化,於是在多方合作努力之下,透過「恩慈專案」為患者做劃時代的治療。手術方式是以如同內視鏡的方式,進行眼內顯微手術加上視網膜下腔注射,從視網膜感光細胞和色素上皮層之間的下間隙注射藥物,該藥物是以腺相關病毒載體帶有正確RPE65的基因,透過注射方式送進視網膜感光細胞內,讓正確的基因序列能持續在眼內不斷製造出正確的蛋白質,改變病程進展。
術後6至8周,男學生的夜盲症狀即獲得改善;術後3個月,眼底和電生理檢查看起來也逐漸正常;半年後視野逐漸擴大;術後一年藥效達到高原期並繼續維持,術後至今兩年半,患者體內藥效發揮的時間與強度都跟國外報告接近,顯示治療結果相當良好。
以前男學生天黑後就無法單獨出門,手術後可以自己走到便利商店買飲料,這對一般人來說再平常不過的事,卻是他前所未有的「創舉」,也照亮了國內眼科基因治療的新時代。男學生的媽媽特地寫信感謝醫療團隊,扭轉了孩子的命運。
LCA的基因治療結果讓人振奮,目前已在申請健保給付中,期待更多患者能夠因此看見希望。目前IRD疾病除了LCA已有基因藥物可治療,另外還有十多種基因藥物陸續研發上市中。
夜盲症是最常見的IRD疾病
在IRD疾病中,以俗稱夜盲症的「視網膜色素病變(retinitis pigmentosa, RP)」最常見,在台盛行率約千分之一,佔IRD病人6成左右。症狀包含夜盲、視覺障礙、對物體明暗對比或顏色分辨能力下降、視野狹窄,可惜尚無基因治療。
至於萊伯氏先天性黑矇症,其盛行率雖只有每十萬名新生兒中約2?3位,但通常一歲之內即會發病,是IRD裡面最早發病的一種,有嚴重視覺功能障礙,常伴隨眼球震顫、眼手徵兆(包括戳眼、壓眼以及揉眼睛等3個特徵),部分患者合併有智能障礙、發展遲緩以及眼球運動失用症。
其他IRD病症還包括錐桿狀細胞失養症(cone-rod dystrophy)、斯特格氏黃斑失養症(Stargardt disease)、以及貝斯特氏卵黃狀黃斑變性(Best disease)等。這些疾病可以在不同年齡發生,並影響到視網膜、黃斑部和脈絡膜等不同的眼睛結構。
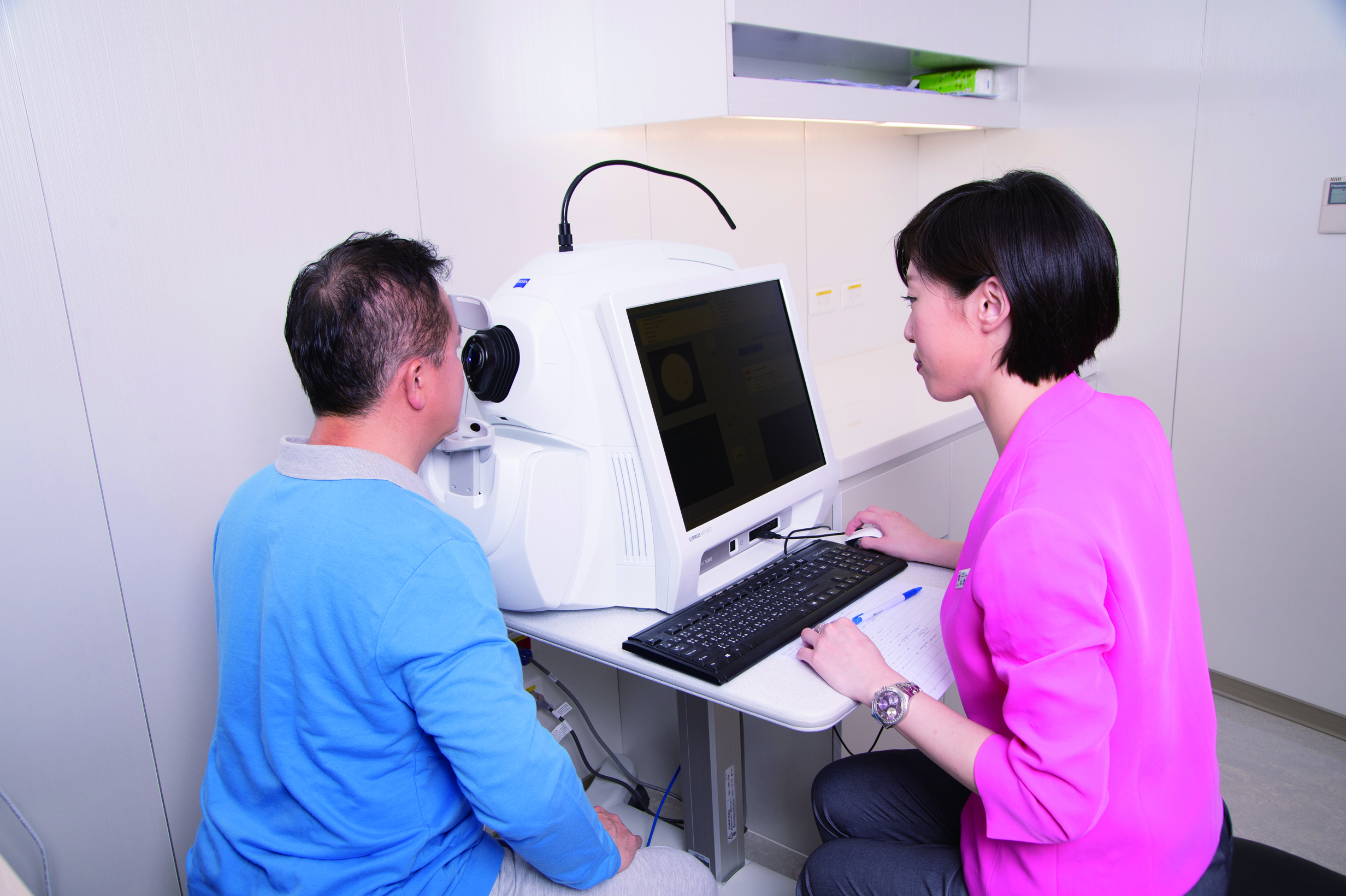
►IRD的病人可透過一系列眼睛檢查診斷疾病,圖為光學電腦斷層掃描儀(OCT)。
雙眼視力同時下降就要提高警覺 注意小小孩是否行動異常
要如何發現IRD呢?由於IRD是基因缺損所導致的先天疾病,與後天因素(如老化、受傷等)造成的視力退化在表現上明顯不同,IRD往往會「對稱性雙眼視力下降」,經常是兩眼同時發病,從眼底影像檢查也看得出「鏡相反應」,即左右眼的退化病徵幾乎像照鏡子,呈現上下左右對稱的表現;若是後天或意外傷害造成的視力退化,通常不會兩眼一起發生。因此,若有雙眼視力同時衰退、夜盲或視野縮小,尤其發生在1、20歲的青少年或者年輕族群時,務必提高警覺,趕快就醫。
如果是3歲之前的小小孩,因為無法做視力測驗,也沒辦法準確表達看到的世界,父母可以從行為觀察孩子是否有視力異常,例如走路常莫名跌倒、常常踢到樓梯或玩具,很可能是視野缺損或縮小所致。
當中心視力變差到一個程度,也可能出現情緒反應遲鈍的情形,例如一般視力正常的小孩看電視會笑,視力差的孩子因為看不清楚,可能沒有反應;或是看到移動的物體也不會像其它小小孩一樣好奇想要去追逐。有些小病人被誤認為是「大雞晚啼、發展遲緩」,其實是視力障礙的緣故。
針對疑似IRD的病人,醫師可能會安排視網膜電流圖 (electroretinography, ERG)、眼電圖(electrooculogram, EOG)或光學電腦斷層掃描儀(OCT)的檢查,以檢測感光細胞和視網膜色素上皮細胞層的功能;螢光眼底血管攝影、暗適應檢查和視野檢查也可以幫助診斷疾病。
儘管基因治療時代來臨,在期待新藥物問世的同時,平日也別忘了「護眼基本功」:作息正常,攝取均衡且充足的營養,尤其眼睛感光細胞需要葉黃素、維生素A和維生素B群等營養素維持健康,並且要減少使用3C產品的時間,從事戶外活動記得戴上太陽眼鏡避免紫外線傷害,盡可能讓視力維持更良好。
基因治療 眼科疾病的新展望
有鑑於IRD是不同基因造成的一大群疾病,2015年起,臺大醫院也執行一項「台灣遺傳性視網膜退化計畫」(Taiwan inherited retinal degeneration project, TIP),並建置「基因性視網膜退化診療平台」,接受全台多處醫療院所的轉診,開始進行大規模臨床診療與基因分析,透過套組檢測作為第一線基因診斷工具,有7成5的病人因此找出缺陷基因,病友可據此作為生涯規劃及是否生育下一代的參考。
這些基因資料庫也是研究本土遺傳性視網膜基因突變流行病學有利的資訊,有助於醫學界了解本土病人輪廓與病情進程,以及接軌可能的新興治療。
隨著基因治療技術日益成熟,未來能運用的範圍也可望會愈來愈廣泛,不只是遺傳性視網膜疾病,目前老年性黃斑部病變也已開始進行基因治療藥物的第二期臨床試驗,這類患者因脈絡膜新生血管產生黃斑部水腫、出血等病症影響視力,每個月都需要定期回診打針,若基因治療成功,直接把藥物注射到視網膜內,藥效可望持續數年之久,減少回診的舟車勞頓。
不過目前基因治療藥價都非常昂貴,期待有朝一日價格變得親民,才不會是看得到用不到的天價藥物,難以普及。

►基因性視網膜退化是先天性疾病,小孩若有雙眼同時視力衰退需提高警覺。
請別錯過本期其他精彩內容…
- 編輯筆記 / 「疫苗」治療癌症 前景可期
- 新聞NEWS一下 / 知名漫畫家大腸癌病逝定期篩檢、避開風險因子 遠離大腸癌
- 新聞NEWS一下 / 知名女星驟逝 生前飽受自體免疫疾病困擾
- 個人化癌症疫苗 開啟癌症治療新頁!
- 研發個人化癌症疫苗 台灣急起直追 奈米疫苗另闢蹊徑
- 肺癌在台灣首度登上發生率第一!篩檢與治療進展迅速 晚期也不用灰心
- 癌症精準醫療跨大步次世代基因定序(NGS)將納健保
- ChatGPT熱潮, 為醫療現場帶來哪些衝擊與影響?
- 人生最後一程如何好好走?「斷食善終」與安寧緩和醫療有什麼不同?
- 健保擴大給付 缺血性腦中風,及時打通血管可免失能!
- 搔癢抓不停、出現黑褐色斑點… 銀髮族常見皮膚問題如何處置?
- 冬天呼吸道病菌齊發!如何治療及照顧?
- 頻尿、漏尿、夜尿… 是間質性膀胱炎嗎?
- 神經有嘻哈 楊智超醫師專欄 / 帶狀疱疹的治療與預防
- 消脂保肝專欄 粘曉菁醫師 / 肥胖加重睡眠呼吸中止症 積極減重可逆轉!
- 曾宇婷醫師專欄 / 睡眠與血糖控制息息相關!
- 透視青光眼 王清泓醫師專欄 / 眼科檢查為什麼需要散大瞳孔?